Functional Neurosurgery · Trainee Resources
Beyond the Box-and-Arrow
Rethinking the Basal Ganglia: Network Physiology and the Levodopa Paradox in DBS
A practical framework for understanding why STN and GPi DBS produce similar motor benefit in Parkinson disease while producing very different medication trajectories.
Orientation
This reading is built around a single clinical observation that most trainees, and most textbooks, never quite resolve: STN DBS routinely cuts levodopa equivalent daily dose by 40–50%. GPi DBS rarely cuts it by more than 20%. Yet motor improvement is roughly equivalent between the two targets, and the Albin-DeLong circuit diagram we all grew up with predicts nothing of the kind.
Learning to sit with that apparent paradox is a useful apprenticeship. The resolution is not a single mechanism; it is three overlapping stories: a clinical-management story, a pathophysiological story, and a modern network/sweet-spot story. Each layer corrects the previous one without replacing it. By the end of this reading, you should be able to explain to a patient why their neurologist is tapering their Sinemet after STN DBS and leaving it largely alone after GPi DBS, and you should be able to predict which future patients will taper well and which will not.
The Mechanistic Disconnect
1.The Apparent Paradox
The classical Albin-DeLong model describes two parallel striatofugal pathways. The direct pathway, from striatum to GPi/SNr, facilitates movement; the indirect pathway, from striatum to GPe to STN to GPi/SNr, inhibits it. Nigral dopamine loss disinhibits the indirect pathway, which drives the STN into a hyperactive, abnormally bursting state, which in turn overdrives the GPi, which in turn over-inhibits the thalamus. Bradykinesia and rigidity follow.
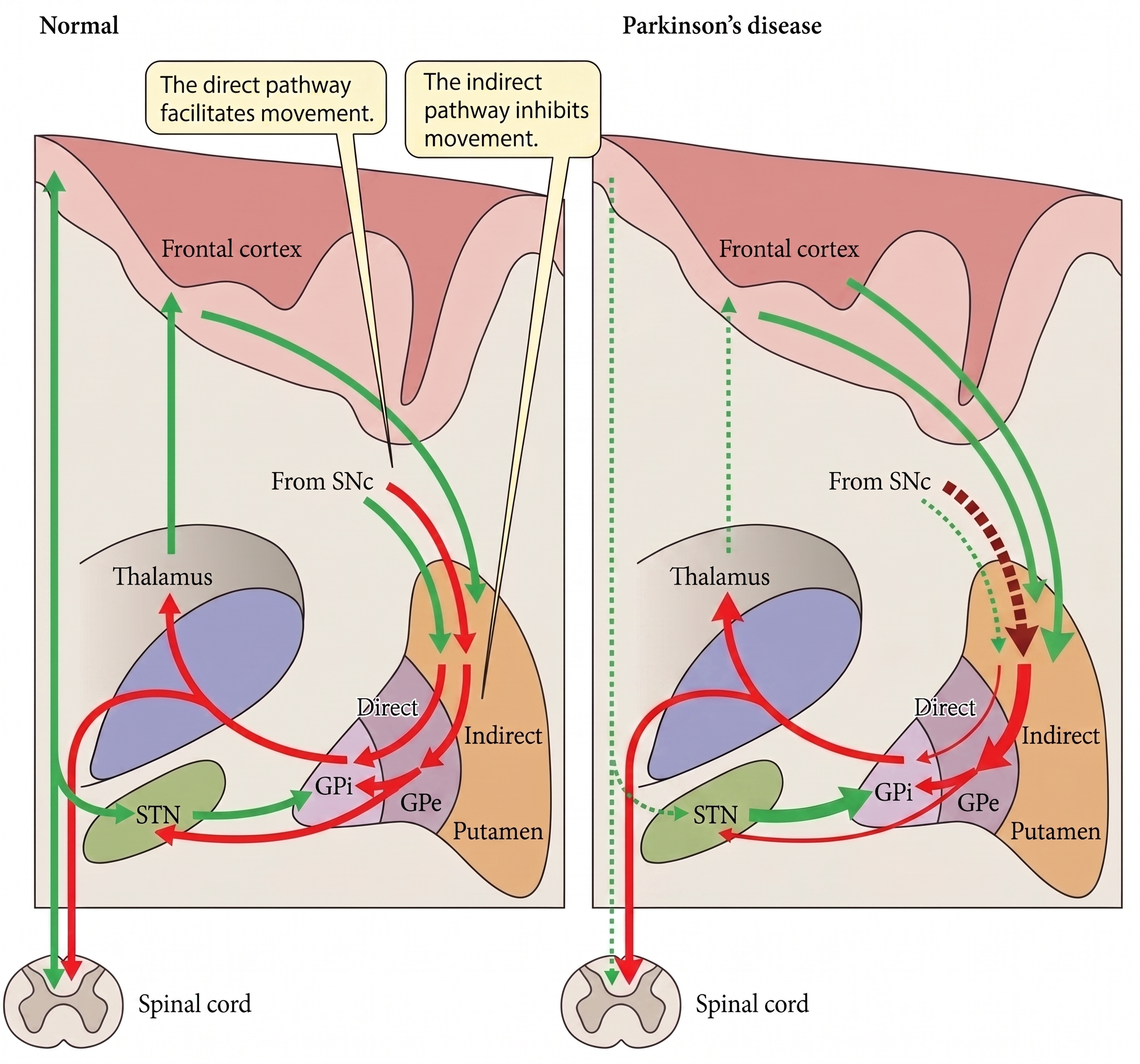
Figure 1.The classical Albin-DeLong rate model. Nigral dopamine loss increases indirect-pathway drive through STN and GPi, producing excessive inhibitory output to thalamus.
Read literally, this schematic tells you that STN and GPi are two points on a serial wire. Silencing either node should produce a comparable downstream effect. The clinical trial data partly support that intuition: motor scores improve similarly at both targets. But then they diverge sharply when you look at medication. In CSP 468, NSTAPS, and essentially every comparative meta-analysis, STN DBS is associated with a substantially greater LEDD reduction, usually 40–50%, while GPi DBS typically reduces LEDD by 0–20%. The schematic does not predict this.
2.First Layer: The Clinical-Practice Answer
The cleanest, most immediately useful framework was articulated by Follett in 2004 and has held up: GPi DBS has a direct anti-dyskinetic effect; STN DBS does not, or only weakly so. This asymmetry drives almost everything downstream about medication management.
After GPi implantation, stimulation itself suppresses dyskinesia regardless of how much levodopa the patient is taking. The neurologist can leave the LEDD essentially unchanged and still get excellent dyskinesia control. There is no mechanical pressure to taper.
After STN implantation, the opposite is true: if you do not reduce levodopa, the patient develops or worsens stim-unmasked dyskinesia. Programming and medication titration are therefore coupled. The team is practically forced to taper the LEDD to get the motor benefit without the chorea. A large fraction of the LEDD-reduction gap between targets is a management artifact, not a distinct pharmacodynamic effect.
A useful shorthand is “ceiling versus floor”: GPi raises the ceiling on how much levodopa a patient can tolerate without dyskinesia; STN raises the floor of motor function so levodopa becomes less necessary. The heuristic is memorable, but be honest about what it hides. It treats the pharmacology as a clinical-management story and says nothing about why STN stimulation substitutes for some of what levodopa was doing. For that we need the pathophysiology.
A practical corollary: work from the Xuanwu group has shown that STN DBS can suppress levodopa-induced on-dyskinesia without any medication reduction, particularly when stimulation is delivered dorsal to the nucleus. So “STN has no direct anti-dyskinetic effect” is too strong. The correct statement is that GPi’s direct effect is larger and more reliable, while STN’s direct effect is real but smaller, contact-dependent, and usually overwhelmed in practice by the need to taper anyway.
3.Second Layer: Where Dopamine Actually Acts
Return to the diagram, and ask a more careful question: where does dopamine physiologically act? The answer is the striatum, not the GPi.
Nigrostriatal dopamine tunes both pathways at the input side of the basal ganglia circuit. D1 drive facilitates the direct pathway, while D2 drive suppresses the indirect pathway. Loss of nigral dopamine propagates down the indirect pathway as pathological STN output. That pathological output is what drives the symptomatic over-inhibition of the thalamus. This is the physiological site of levodopa’s action.
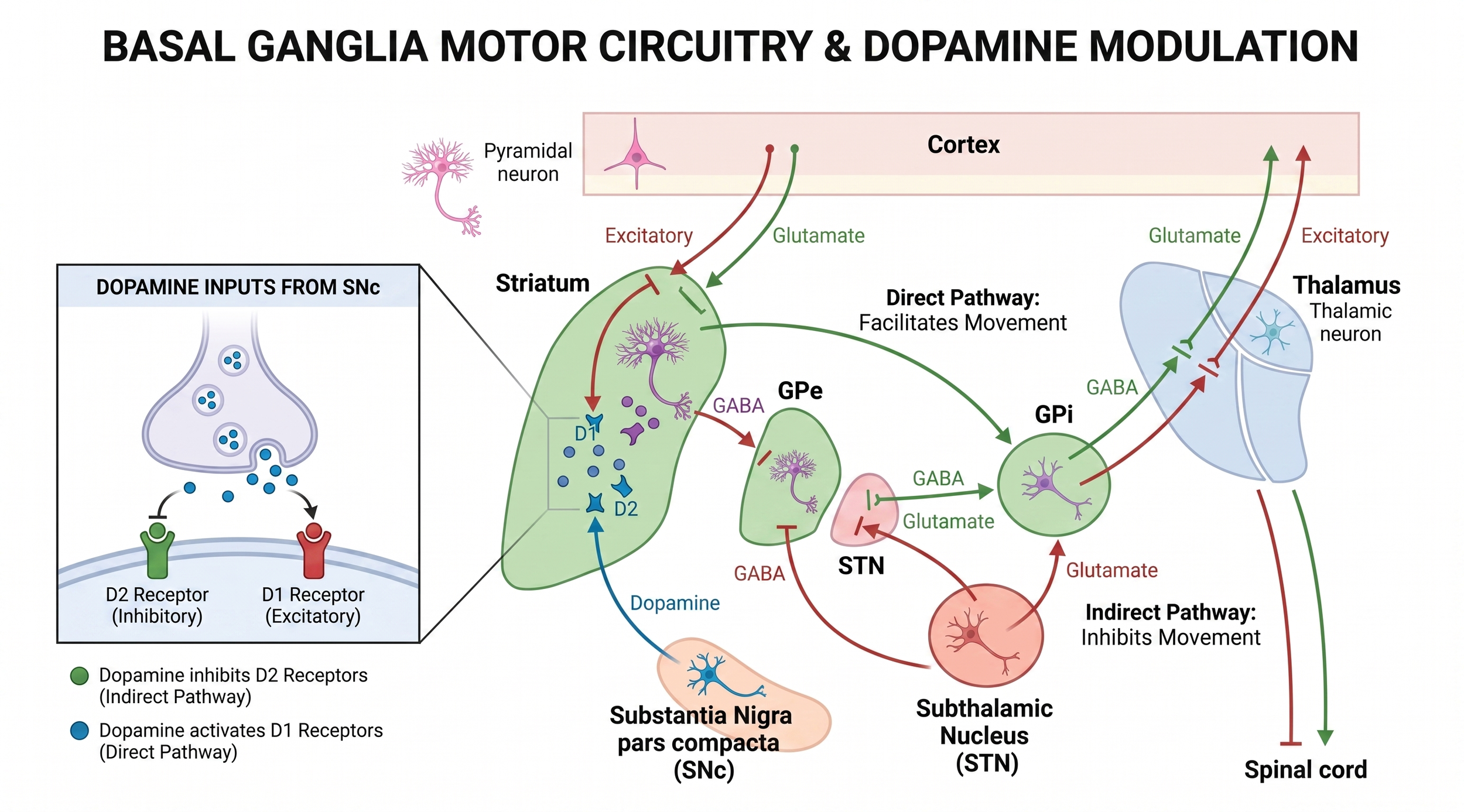
Figure 2.Where dopamine acts. Dopamine modulates the basal ganglia at the striatal input stage, so STN and GPi stimulation occupy different positions relative to levodopa physiology.
Now look at where each DBS target sits relative to that physiology. STN DBS sits at the node dopamine was normally regulating through the indirect pathway. Suppressing or replacing the pathological STN signal effectively substitutes for part of what dopaminergic tone was doing. There is a real physiological overlap between what levodopa does and what STN stimulation does, even though the proximate mechanism differs: D2 receptor action in the striatum versus electrical modulation of STN neurons and their afferents and efferents. That overlap is the reason patients can take less drug and still function.
GPi DBS sits one station further downstream, at the output of the basal ganglia. It masks or overrides the final common output regardless of what is feeding in. The upstream pathological drive is still there; you have just silenced its expression at the gate. There is no substitution for dopaminergic function. The striatum still wants its dopamine, and so there is no physiological room to cut the drug.
A third mechanism supports this framing. STN neurons send collaterals to the substantia nigra pars compacta, and PET imaging with the VMAT2 tracer [¹¹C]-DTBZ has shown changes in striatal dopamine-related binding after STN DBS that are consistent with enhanced release from surviving nigral neurons. GPi DBS, lacking this direct feedback onto the SNc, cannot do this. It is small quantitatively, but it is a real dopamine-sparing effect that only STN stimulation possesses.
4.Third Layer: The Modern Network View
The Albin-DeLong model was built on firing rates. The modern view is built on firing patterns, specifically pathological synchrony in the beta band, roughly 13–30 Hz, across the cortico-basal ganglia-thalamic loop. In Parkinson disease, beta oscillations dominate the STN local field potential, and their amplitude scales with bradykinesia and rigidity. Dopaminergic replacement suppresses them. High-frequency STN DBS suppresses them too. The convergence is not accidental.
The hyperdirect pathway. Oswal and colleagues used simultaneous MEG and STN-LFP recordings with structural tractography to show that high-beta coherence between the supplementary motor area and STN correlates directly with hyperdirect pathway fiber density, and that cortex drives STN at these frequencies, not the other way around. The sweet spot for STN DBS, the dorsolateral border of the nucleus and adjacent white matter, is exactly where these hyperdirect fibers enter. Akram et al. mapped the cortical fingerprint of effective STN stimulation and showed that tractographic connectivity to M1 predicts tremor improvement, to SMA predicts bradykinesia improvement, and to both SMA and prefrontal cortex predicts rigidity improvement. The best current mechanistic account is that STN DBS works in large part by antidromic modulation of cortico-STN fibers, disrupting pathological cortex-to-STN beta coupling at its source.
ERNA and the indirect pathway. Evoked resonant neural activity, the stimulation-locked high-frequency oscillation that appears in the STN during DBS, is now understood as a signature of DBS-mediated engagement of the GPe-STN indirect-pathway loop. ERNA localization predicts motor outcome, and it is engaged by both STN and GPi stimulation. This is probably part of why GPi DBS works at all despite being downstream of dopamine’s site of action, and why both targets produce similar motor improvement despite their different relationships to the drug.
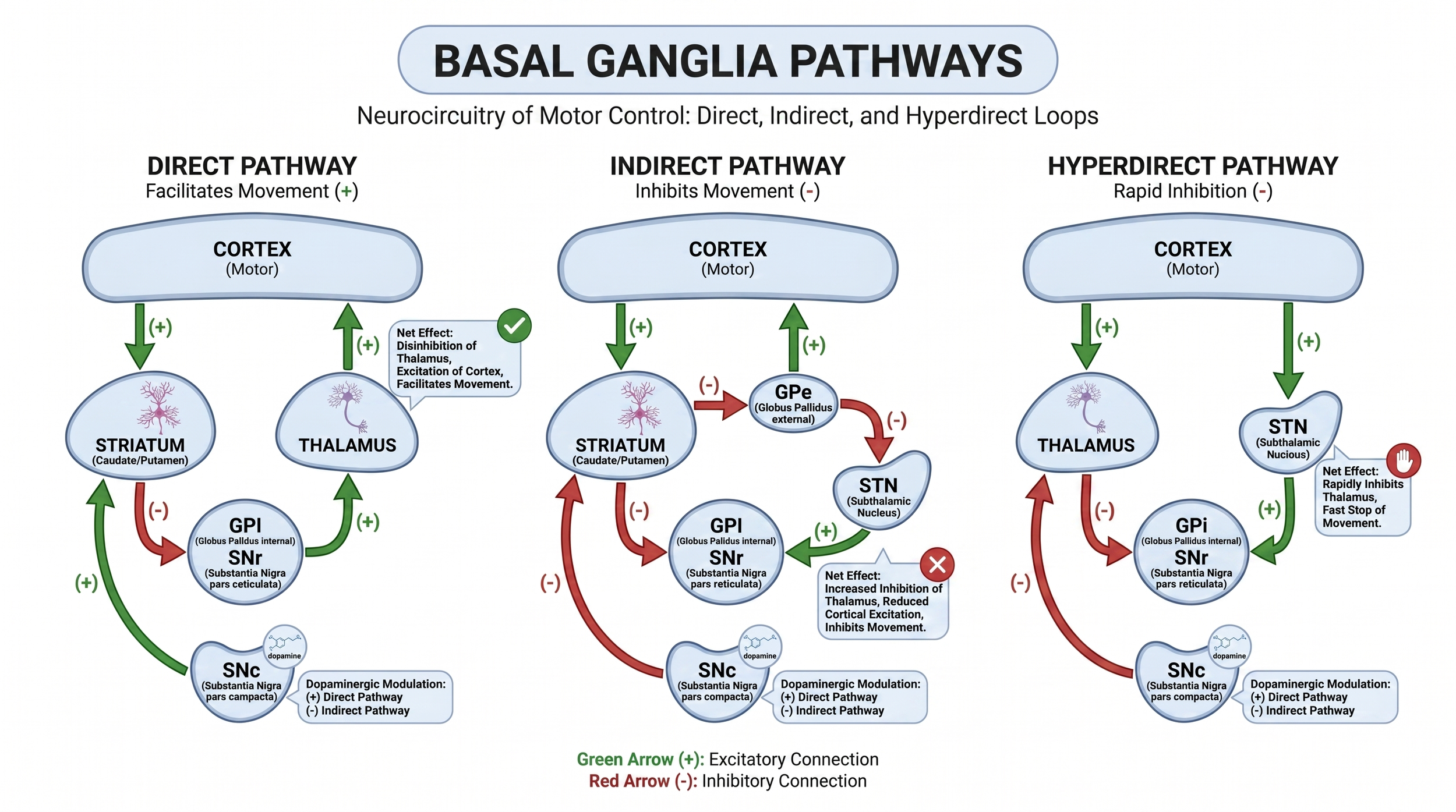
Figure 3.The three cortical-to-basal-ganglia pathways. Direct, indirect, and hyperdirect pathways explain why STN DBS is better understood as network modulation than as simple node inhibition.
Two practical implications follow. First, “STN DBS mimics levodopa” is a useful teaching line but mechanistically imprecise. They converge on the same physiological signature, suppression of pathological beta synchrony, via different molecular mechanisms. Second, GPi DBS does not achieve the same cortex-STN beta suppression via the hyperdirect pathway, which is part of why it cannot substitute for the drug.
Clinical Application and Target Selection
5.Good Lead Placement Does Not Equal Big LEDD Reduction
This is the advanced nugget, and it is the one most clinically useful to carry into the OR and the programming clinic. Motor improvement and LEDD reduction are not the same outcome, and they do not scale together. They have partially different predictors.
The Dembek 2019 Annals of Neurology paper quantified how much motor improvement comes from hitting the correct anatomical target. Overlap of the volume of tissue activated with the probabilistic sweet spot, located at the dorsolateral border of the STN and extending into adjacent white matter, explained R² = 37% of the variance in motor improvement. That was more than triple what was explained by raw STN coverage or sensorimotor STN coverage. In short: lead placement matters enormously for motor outcome, and centroid-of-STN is a weaker predictor than sweet-spot overlap.
Now the Benoit-Marand mediation analysis: 144 patients, Boruta feature selection, and a structural equation model to separate direct effects on LEDD reduction from motor-mediated effects. Mean LEDD reduction was 41.7%, and mean motor improvement was 48.6%. Three predictors were directly associated with lower LEDD reduction, independent of motor improvement: lower baseline LEDD, greater axial impairment, and higher total volume of tissue activated. Sensorimotor STN overlap was positively associated with motor improvement but not with LEDD reduction.
This is the mechanistic argument for directional leads, image-guided programming, and tractography-informed contact selection. It is also the argument against promising a patient any specific LEDD reduction preoperatively. A patient with heavy axial disease or relatively low baseline LEDD will not drop their medications by 50% no matter how good the lead placement is, because the biology will not allow it. That is not a failure of technique. It is a separate axis asserting itself.
6.What the Comparative Trials Actually Show
Four studies form the spine of the evidence base. Read at least the first three in full before lecture.
Limousin 1995. The founding three-patient series of bilateral STN DBS for advanced Parkinson disease. Activities of daily living improved 58–88%; motor scores improved 42–84%. This is where the field starts, and the paper is short enough to read in fifteen minutes.
CSP 468 / Follett 2010. 299 patients, VA-NINDS multicenter, randomized STN versus GPi. The primary outcome, off-medication, on-stimulation UPDRS-III, did not differ significantly between targets. Patients in the STN arm required significantly lower dopaminergic doses. STN patients had a greater decline in one component of processing speed; depression worsened after STN and improved after GPi. This is the trial that made “target selection should be individualized” an evidence-based statement rather than a prejudice.
NSTAPS / Odekerken 2013. Dutch multicenter RCT, 128 patients. Hypothesis: GPi stimulation would produce greater functional improvement than STN because of fewer cognitive and mood complications. The data contradicted the hypothesis. STN was better on off-period motor improvement and disability, dyskinesia reduction was larger with GPi, and LEDD reduction was significantly greater with STN. Three-year follow-up confirmed the motor advantage and the absence of a cognitive safety penalty for STN.
CSP 468F / Ostrem 2025. Ten-year extension of CSP 468. Motor improvement was sustained in both arms. The LEDD reduction gap between targets narrows considerably at very long follow-up. Useful to close the loop with, because it reminds trainees that “STN spares medications more than GPi” is largely a short-to-intermediate-term phenomenon; disease progression drives both groups toward similar LEDD trajectories over a decade.
7.Target Selection Framework
Given the two targets are equivalent for motor improvement, selection is driven almost entirely by the patient’s profile of symptoms, side effects, cognitive reserve, and treatment goals.
Favors STN: younger patient with intact cognition and mood; medication burden is the problem, including impulse control disorder, dopamine dysregulation syndrome, dopa-induced nausea, or medication-induced hallucinations; battery longevity or economic considerations favor the lower stimulation amplitudes typical of STN; severe off-period bradykinesia or rigidity; need for more off-period motor improvement.
Favors GPi: dyskinesia-dominant disease with heavy reliance on levodopa that you do not want to taper; baseline cognitive vulnerability or mild cognitive impairment; depression or emotional lability at baseline; older or more advanced patient; need for programming flexibility; anticipated unilateral implantation; surgeon or team earlier on the learning curve, because GPi is more forgiving.
Both targets fail: axial symptoms, including gait freezing, postural instability, and speech, and non-levodopa-responsive symptoms. The levodopa challenge is your most reliable predictor of DBS motor outcome. If a symptom does not respond to dopamine, it will not reliably respond to stimulation at either target. Setting this expectation preoperatively is half the battle.
8.Pearls for the Operating Surgeon
The target is the dorsolateral STN and adjacent white matter, not the center of the nucleus. “Nice lead through the middle of STN on post-op MRI” is a weaker predictor of outcome than “VTA overlapping the sweet spot and the hyperdirect pathway entry zone.” Plan accordingly.
A smaller, focused VTA beats a larger one. Bigger stimulation fields recruit non-motor STN territory and its side effects, narrowing the therapeutic window and capping how aggressively the neurologist can cut medications. Directional leads earn their cost here.
Motor outcome and LEDD reduction are separate axes with different predictors. Do not promise a patient a specific LEDD drop preoperatively. Promise motor improvement contingent on levodopa responsiveness, and discuss medication management as a separate question that depends on baseline biology and how the team co-manages stimulation and drug.
The forced taper is therapeutic, not incidental. For patients with dopamine dysregulation syndrome, severe impulse control disorders, or dopamine-induced hallucinations, STN DBS is the right answer specifically because the medication reduction is itself part of the treatment. Say this explicitly in clinic.
Pearls
- STN and GPi improve motor scores similarly, but STN reduces LEDD much more in the short-to-intermediate term.
- The LEDD difference is partly clinical management: GPi directly suppresses dyskinesia, so there is less pressure to taper medication.
- The LEDD difference is partly physiology: STN sits at the node dopamine normally regulates through the indirect pathway.
- STN DBS and levodopa converge on suppression of pathological beta synchrony, but by different mechanisms.
- GPi DBS gates the final common output without substituting for striatal dopaminergic tone.
- Motor improvement and LEDD reduction are different outcomes with different predictors.
- Sweet-spot overlap predicts motor improvement; contained VTA and patient-level biology predict medication tapering room.
- Do not promise a specific LEDD reduction. Counsel medication reduction as a separate, biologically constrained outcome.
- Favor STN when medication burden is the problem. Favor GPi when dyskinesia, cognition, mood, or programming flexibility dominates.
- Read Limousin 1995. Four pages, three patients, and the beginning of the modern STN DBS era.
Must-Read References
- Limousin P, Pollak P, Benazzouz A, et al. Effect of parkinsonian signs and symptoms of bilateral subthalamic nucleus stimulation. Lancet. 1995;345(8942):91–95. The founding STN DBS paper.
- Follett KA, Weaver FM, Stern M, et al.; CSP 468 Study Group. Pallidal versus subthalamic deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2010;362(22):2077–2091. The central randomized comparison for target selection.
- Odekerken VJ, van Laar T, Staal MJ, et al. Subthalamic nucleus versus globus pallidus bilateral deep brain stimulation for advanced Parkinson’s disease: a randomised controlled trial. Lancet Neurol. 2013;12(1):37–44. The NSTAPS trial.
- Ostrem JL, Luo P, Weaver FM, et al.; CSP 468F Study Group. 10-year clinical outcomes of subthalamic nucleus versus pallidal deep brain stimulation for Parkinson’s disease: VA/NINDS CSP #468F. Front Neurol. 2025;16:1728999. The long-view follow-up.
- Follett KA. Comparison of pallidal and subthalamic deep brain stimulation for the treatment of levodopa-induced dyskinesias. Neurosurg Focus. 2004;17(1):E3. The clean clinical explanation for the dyskinesia asymmetry.
- Dembek TA, Roediger J, Horn A, et al. Probabilistic sweet spots predict motor outcome for deep brain stimulation in Parkinson disease. Ann Neurol. 2019;86(4):527–538. Why sweet-spot overlap beats centroid thinking.
- Benoit-Marand M, et al. Understanding dopaminergic dose reduction following STN-DBS: mediation analysis. J Neurol Neurosurg Psychiatry. 2026. The key modern paper separating motor improvement from LEDD reduction.
- Akram H, Sotiropoulos SN, Jbabdi S, et al. Subthalamic deep brain stimulation sweet spots and hyperdirect cortical connectivity in Parkinson’s disease. NeuroImage. 2017;158:332–345. The connectomic bridge between sweet spot and hyperdirect pathway.